Налтрексон
Содержание:
- Пенициллины
- Биологические барьеры организма
- Физико-химические свойства
- Хлорамфеникол
- § 39. Закон радиоактивного распада
- Литература
- Тетрациклины
- Период полувыведения
- Токсикологические свойства и характеристики
- Примеры значений и уравнений
- Действие на вредные организмы
- Выведение лекарств из организма
- Период полураспада — это просто
- Макролиды
- Период полувыведения лекарства это: возраст и лекарства – Помощь Медика
- Фторхинолоны
- Итоги
Пенициллины
Основным современным представителем пероральных пенициллинов является амоксициллин. Его наиболее характерные фармакокинетические свойства :
- Сохраняет активность в очаге воспаления (при низком pH, высокой концентрации лейкоцитов, белка);
- Хорошо всасывается из ЖКТ, что объясняет эффективность пероральной формы: абсорбция составляет 75–93 %;
- Биодоступность не зависит от пищи, то есть можно применять и до, и после еды;
- В небольшом количестве проникает в грудное молоко, однако не оказывает негативного действия на младенцев (за исключением риска сенсибилизации, диареи и кандидоза слизистой полости рта);
- Проникает через плацентарный барьер, но не оказывает негативного влияния на плод и фертильность. Безопасен при беременности, поэтому широко применяется в этот период, «если потенциальная польза для матери превышает потенциальный риск для плода»;
- Плохо проникает через гематоэнцефалический барьер;
- Как и все пенициллины, очень быстро выводится почками.
Биологические барьеры организма
Большинство препаратов легко преодолевает стенку капилляров. Одни средства проникают через поры путем фильтрации, другие проникают через капиллярную стенку путем диффузии. Некоторые гидрофильные соединения преодолевают капиллярную стенку с помощью транспортных систем.
Гематоэнцефалический барьер является существенным препятствием на пути проникновения лекарств в ЦНС. Капилляры мозга не имеют пор, в них отсутствует пиноцитоз. Кроме того, внешняя поверхность эндотелия сосудов выслана астроглией, что создает дополнительный барьер на пути препаратов в ЦНС. В общем, гидрофильные соединения плохо проникают в мозг, а липофильные — хорошо. Во время воспалительных процессов мозговых оболочек проницаемость ГЭБ увеличивается.
Физико-химические свойства
Химически чистый дикват дибромид – белые гигроскопические кристаллы. Соединение хорошо растворимо в воде, плохо в спиртах. В неполярных органических растворителях практически нерастворимо. По данным хорошо растворяется в воде и спиртах.
Вещество в щелочной среде легко гидролизуется, в кислом и нейтральном растворах стабильно. Под действием УФ-облучения разлагается (период полураспада составляет менее недели).
Длительно сохраняется в полиэтиленовых канистрах, корродирует металлы (сталь, жесть, оцинкованную жесть).
Выпускается в виде 15% водного раствора соли брома (в пересчете на катион).
Катион является биологически активной частью молекулы. Водные растворы нелетучи, под действием сильных щелочей образуют окрашенные комплексные продукты.
Хлорамфеникол
Также отдельно стоит рассмотреть фармакокинетику хлорамфеникола :
- Хлорамфеникол хорошо всасывается из пищеварительного тракта, биодоступность составляет 70–80 %;
- Всасываемость и биодоступность не зависит от приема пищи;
- Хорошо распределяется в организме, создавая высокие концентрации в бронхиальном секрете, желчи, а также в цереброспинальной жидкости и головном мозге даже при отсутствии менингиального воспаления;
- Проникает через гематоэнцефалический барьер;
- Выделяется через почки, причем в неактивном состоянии. Поэтому при почечной недостаточности необходимости в снижении дозы нет.
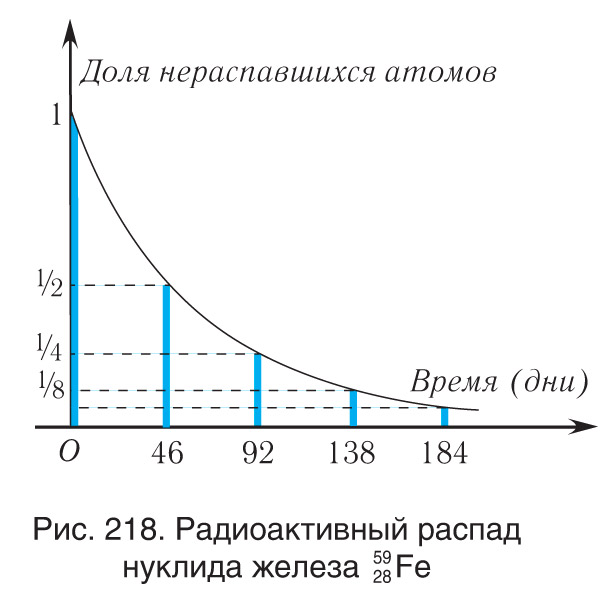
§ 39. Закон радиоактивного распада
|
При всем разнообразии реакций самопроизвольного (спонтанного) распада ядер в этом процессе наблюдается общая закономерность, которую можно описать математически. Интересно, что зависимость количества распавшихся ядер от времени задается одной и той же функцией для различных ядер, участвующих в распаде. Перейдем к количественному описанию процессов радиоактивного распада. |
Большинство изотопов любого химического элемента превращается в более устойчивые изотопы путем радиоактивного распада. Каждый радиоактивный элемент распадается со своей, присущей только ему «скоростью». При этом для каждого радиоактивного ядра существует характерное время, называемое периодом полураспада , спустя которое в исходном состоянии остается половина имевшихся ядер. Таким образом, периодом полураспада называется промежуток времени, за который распадается половина начального количества радиоактивных ядер. Другая половина ядер превращается в более устойчивые изотопы посредством распада.Отметим, что период полураспада не зависит от того, в каком состоянии находится вещество: твердом, жидком или газообразном. Кроме того, период полураспада радиоактивного вещества не зависит от его количества, от времени, места и условий, в которых оно находится. Поэтому количество радиоактивных ядер «тогда» и «сейчас» непосредственно определяет промежуток времени , прошедший с момента уменьшения числа ядер от до .Невозможно точно предсказать, когда произойдет распад данного ядра. Однако можно оценить среднее число ядер, которые распадутся за данный промежуток времени. Таким образом, закон радиоактивного распада является статистическим и он справедлив только при достаточно большом количестве радиоактивных ядер.
 |
 |
Для записи закона радиоактивного распада будем считать, что в начальный момент времени () число радиоактивных ядер . Через промежуток времени, равный периоду полураспада, это число будет , еще через такой же промежуток времени — (рис. 218). Спустя промежуток времени, равный n периодам полураспада , радиоактивных ядер останется:
|
|
(1) |
Это соотношение выражает закон радиоактивного распада, который можно сформулировать следующим образом:
число нераспавшихся радиоактивных ядер убывает с течением времени по закону, представленному соотношением (1).
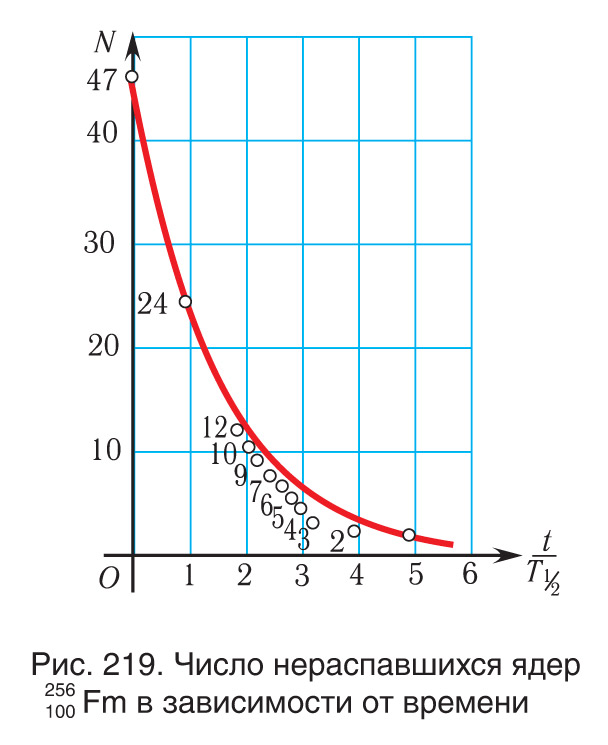
Закон радиоактивного распада позволяет найти число нераспавшихся ядер в любой момент времени. Полученное выражение хорошо описывает распад радиоактивных ядер, если их количество достаточно велико.Приведем экспериментальные результаты, которые показывают, что при малом количестве радиоактивных ядер это выражение неприменимо. На рисунке 219 изображен график распада 47 ядер изотопа фермия , период полураспада которого . Из рисунка 219 видно, что пока ядер было достаточно много — от 47 до 12, то показательная функция хорошо описывала закон распада. Однако при меньшем числе ядер истинная зависимость существенно отличается от показательной функции.Периоды полураспада некоторых радиоактивных изотопов веществ приведены в таблице 11.
| Таблица 11. Периоды полураспада радиоактивных изотопов веществ | |
| Вещество | Период полураспада |
| 30,17 лет | |
| 5,3 года | |
| 8,04 суток | |
| 24 390 лет | |
| 1600 лет | |
| 3,8 суток | |
| 700 млн лет | |
| 4,5 млрд лет |
|
Впервые процесс радиоактивного распада для измерения промежутков времени был использован в 1904 г. Резерфордом. По отношению концентрации урана и его дочернего продукта распада (гелия) он определил возраст урановой породы. Эта работа положила начало ядерной геохронологии — определению возраста различных минералов Земли по радиоактивным долгоживущим веществам. В дальнейшем исследование процессов ядерного синтеза позволило перейти к ядерной космохронологии, т.е. к определению продолжительных промежутков времени, прошедших с момента образования элементов в масштабах Галактики и Вселенной. В основу ядерной космохронологии положена неизменность «скорости» радиоактивного распада. В 1927 г. американский ученый Г. Блюмгарт, используя изотоп , впервые определил скорость кровотока у людей. В 1934 г. венгерский ученый Дьердь фон Хевеши, используя дейтерий, впервые установил, что в организме человека вода полностью обновляется в течение 14 суток. В 1943 г. Дьердь фон Хевеши была присуждена Нобелевская премия по химии «за работу по использованию изотопов в качестве меченых атомов при изучении химических процессов». |
 |
Литература
- Donald R.A. Uges TIAFT Bulletin of the international Association of Forensic Toxicologist, VOLUME 26 number 1 SUPPLEMENT. 1996.
- Winek C.L. Drug and chemical blood-level data 1994 in Winek»s Toxicological Annual, 1994.
- Donald R.A. Uges, Response to Compilation of Therapeutic Drug Levels, Letter to the Editor, TIAFT Bulletin 1996, 26(1):18-19.
- Jones G. to Compilations of Therapeutic Drug Levels, Letter to the Editor, TIAFT Bulletin 1995; 25 (4) : 15-18.
Перевод на русский язык осуществлен Р.Р.Красновой (Республиканский центр судебно-медицинской экспертизы МЗ и МП РФ, директор проф. Томилин).
Подготовлено к печати Липецким областным бюро судебно-медицинской экспертизы, начальник Флейшер М.Р.
Тетрациклины
Общие черты фармакокинетики препаратов этой группы выглядит следующим образом :
- Биодоступность доксициклина составляет 90–100 % и практически не зависит от приема пищи, тетрациклина — 75 % натощак и около 37 % при приеме с пищей. Соответственно, тетрациклин нужно принимать натощак;
- Все тетрациклины связываются и инактивируются двухвалентными катионами, в том числе кальцием и магнием. Поэтому совместный прием антибиотиков этой группы и препаратов кальция или магния влияет на эффективность тетрациклинов. Чтобы избежать лекарственных взаимодействий, антимикробные препараты нужно принимать натощак;
- Распределяются во многих органах и тканях. Доксициклин по сравнению с тетрациклином создает более высокие тканевые концентрации;
- Проникают через плацентарный барьер и в грудное молоко в высоких концентрациях. Противопоказаны при беременности и лактации;
- Метаболизируются в печени;
- Тетрациклин выводится преимущественно почками, поэтому при почечной недостаточности необходима коррекция дозы. Доксициклин выводится почками и через ЖКТ, причем при поражении почек пищеварительный путь выведения становится основным;
- Период полувыведения доксициклина в два раза выше по сравнению с тетрациклином — 15–24 часа и 8 часов, соответственно.
Период полувыведения
Процесс очистки организма от медицинского вещества путем его инактивации называется «элиминацией». Для оценки элиминации используют величину, которая называется «период полувыведения» (Т 1/2). Период элиминации — это время, за которое концентрация в крови уменьшается вдвое в сравнении с фазой равновесного распределения.
Необходимо отметить, что с увеличением дозы препарата выведение его из организма снижается и соответственно возрастает период полувыведения.
Кроме того, для количественной характеристики скорости вывода вещества из организма используют термин «клиренс» (очищение). Он отражает скорость очистки плазмы крови от вещества (например, 10 мл / мин). Различают общий, почечный и печеночный клиренс.
Большинство лекарственных средств несут в организм метаболические изменения. Этот процесс называется биотрансформацией. Суть метаболических превращений заключается в том, чтобы чужеродное, опасное для организма средство превратилось в соединение, которое может быть легко выведено с мочой, желчью или потом. Такие полярные метаболиты плохо растворяются в липидах и имеют низкую способность взаимодействовать с белками плазмы крови и тканей. Метаболиты, как правило, плохо проникают через биологические мембраны и не испытывают реабсорбции в почках и кишечнике.
Метаболизм лекарств в организме
Метаболизм лекарственных средств происходит преимущественно в микросомальном аппарате печени. Некоторые метаболические преобразования определенных лекарств могут происходить в кишечнике, легких, коже и плазме крови. Лишь некоторые препараты выводятся из организма в неизмененном виде.
Известны два базовых вида метаболизма ЛС:
- метаболическую трансформацию (МТ);
- конъюгации.
Окисление — один из самых распространенных путей инактивации лекарств. Окисление последних происходит в печени с участием микросомальных ферментов оксидаз (основной представитель цитохром Р-450). Суть окисления заключается в отщеплении ионов водорода от боковых цепей молекул препаратов. В реакции участвуют НАДФ и кислород.
Восстановление является более редким путем метаболизма лекарств. Реакции восстановления катализируют такие ферментные системы, как нитро- и азоредуктазы и др.
Конъюгации — это реакция присоединения к молекуле ЛС определенного гидрофильного эндогенного метаболита. Эти метаболиты предварительно активируются, образуя макроэргическую связь за счет АТФ. Типичной реакцией конъюгации является присоединение к молекулам препаратов остатков уксусной или глюкуроновой кислот, глутатиона, сульфатов, глицина, метильного остатка и др. Конъюгация может быть единственным путем преобразования лекарственных веществ в организме, или же она происходит после предварительной МТ. В процессе МТ и конъюгации препараты, как правило, теряют биологическую активность.
Некоторые препараты могут подавлять микросомальные ферменты печени (левомицетин, бутадион и др.) или немикросомальные ферменты (антихолинэстеразные средства, ингибиторы МАО и др.). В таких случаях действие лекарств, метаболизм которых происходит при участии соответствующих ферментов, увеличивается. В то же время существуют соединения (фенобарбитал и др.), которые повышают (индуцируют) скорость синтеза микросомальных ферментов.
Токсикологические свойства и характеристики
В почве
Поскольку дикват представляет собой катион, то при попадании в почву он быстро инактивируетя благодаря связыванию с отрицательно заряженными почвенными частицами. В связи с этим дикват почти не передвигается в почве, не вымывается из нее водой и не всасывается корнями растений, а вскоре после внесения не оказывает гербицидного действия.
На поверхности почвы быстро разрушается под действием ультрафиолетовых лучей.
Теплокровные
ЛД50
ЛД50 при пероральном введении для крыс и кроликов – 281,9-227,6, для мышей – 79,8, при кожной аппликации для кроликов – 400 мг/кг. Обладает выраженной кожно-резорбтивной токсичностью: ЛД50 для крыс 726,9, раздражает кожу и слизистые оболочки дыхательных путей, вызывает конъюктивит. Кумулятивные свойства выражены слабо.
Препарат оказывает влияние на окислительные процессы, изменяя активность каталазы и пероксидазы крови.
Не обнаружено канцерогенное воздействие при скармливании крысам на протяжении 14 месяцев пищи, содержащей дикват в количестве 500 мг/кг.
Примеры значений и уравнений
| Характерная черта | Описание | Условное обозначение | Ед. изм | Формула | Рабочий пример значения |
|---|---|---|---|---|---|
| Доза | Количество введенного препарата. | D{\ displaystyle D} | мол{\ Displaystyle \ mathrm {mol}} | Расчетный параметр | 500 ммоль |
| Интервал дозирования | Время между введениями дозы препарата. | τ{\ Displaystyle \ тау} | s{\ Displaystyle \ mathrm {s}} | Расчетный параметр | 24 ч |
| C макс. | Пиковая концентрация препарата в плазме после приема. | CМаксимум{\ displaystyle C _ {\ text {max}}} | M{\ displaystyle \ mathrm {M}} | Прямое измерение | 60,9 ммоль / л |
| t макс | Время достижения C макс . | тМаксимум{\ Displaystyle т _ {\ текст {макс}}} | s{\ Displaystyle \ mathrm {s}} | Прямое измерение | 3.9 ч |
| C мин | Низкая ( корыта ) концентрация , что лекарственное средство достигает до следующей дозы вводят. | Cмин,SS{\ displaystyle C _ {{\ text {min}}, {\ text {ss}}}} | M{\ displaystyle \ mathrm {M}} | Прямое измерение | 27,7 ммоль / л |
| Объем распространения | Кажущийся объем, в котором распределено лекарство (т.е. параметр, связывающий концентрацию лекарства в плазме с количеством лекарства в организме). | Vd{\ displaystyle V _ {\ text {d}}} | м3{\ Displaystyle \ mathrm {м} ^ {3}} | DC{\ displaystyle {\ frac {D} {C_ {0}}}} | 6.0 л |
| Концентрация | Количество препарата в заданном объеме плазмы . | C,CSS{\ displaystyle C_ {0}, C _ {\ text {ss}}} | M{\ displaystyle \ mathrm {M}} | DVd{\ displaystyle {\ frac {D} {V _ {\ text {d}}}}} | 83,3 ммоль / л |
| Период полураспада | Время, необходимое для всасывания в системный кровоток 50% заданной дозы препарата. | т12а{\ displaystyle t _ {{\ frac {1} {2}} а}} | s{\ Displaystyle \ mathrm {s}} | пер(2)kа{\ Displaystyle {\ гидроразрыва {\ ln (2)} {к _ {\ текст {а}}}}} | 1.0 ч |
| Константа скорости абсорбции | Скорость, с которой лекарство попадает в организм при пероральном и других внесосудистых путях. | kа{\ Displaystyle к _ {\ текст {а}}} | s-1{\ Displaystyle \ mathrm {s} ^ {- 1}} | пер(2)т12а{\ Displaystyle {\ гидроразрыва {\ ln (2)} {т _ {{\ гидроразрыва {1} {2}} а}}}} | 0,693 -1 |
| Период полувыведения | Время, необходимое для того, чтобы концентрация препарата достигла половины исходного значения. | т12б{\ displaystyle t _ {{\ frac {1} {2}} b}} | s{\ Displaystyle \ mathrm {s}} | пер(2)kе{\ Displaystyle {\ гидроразрыва {\ ln (2)} {к _ {\ текст {е}}}}} | 12 часов |
| Константа скорости элиминации | Скорость, с которой лекарство выводится из организма. | kе{\ displaystyle k _ {\ text {e}}} | s-1{\ Displaystyle \ mathrm {s} ^ {- 1}} | пер(2)т12бзнак равноCLVd{\ displaystyle {\ frac {\ ln (2)} {t _ {{\ frac {1} {2}} b}}} = {\ frac {CL} {V _ {\ text {d}}}}} | 0,0578 ч -1 |
| Скорость инфузии | Скорость инфузии, необходимая для баланса выведения. | kв{\ displaystyle k _ {\ text {in}}} | молs{\ Displaystyle \ mathrm {моль / с}} | CSS⋅CL{\ displaystyle C _ {\ text {ss}} \ cdot CL} | 50 ммоль / ч |
| Площадь под кривой | Интеграл кривой концентрация-время (после однократной дозы или в стационарном состоянии). | АUC-∞{\ displaystyle AUC_ {0- \ infty}} | M⋅s{\ Displaystyle \ mathrm {M} \ cdot \ mathrm {s}} | ∫∞Cdт{\ displaystyle \ int _ {0} ^ {\ infty} C \, \ operatorname {d} t} | 1320 ммоль / л · ч |
| АUCτ,SS{\ displaystyle AUC _ {\ tau, {\ text {ss}}}} | M⋅s{\ Displaystyle \ mathrm {M} \ cdot \ mathrm {s}} | ∫тт+τCdт{\ displaystyle \ int _ {t} ^ {t + \ tau} C \, \ operatorname {d} t} | |||
| Оформление | Объем плазмы, очищенной от препарата за единицу времени. | CL{\ displaystyle CL} | м3s{\ Displaystyle \ mathrm {м} ^ {3} / \ mathrm {s}} | Vd⋅kезнак равноDАUC{\ displaystyle V _ {\ text {d}} \ cdot k _ {\ text {e}} = {\ frac {D} {AUC}}} | 0,38 л / ч |
| Биодоступность | Системно доступная фракция лекарственного средства. | ж{\ displaystyle f} | Безразмерный | АUCпо⋅DivАUCiv⋅Dпо{\ displaystyle {\ frac {AUC _ {\ text {po}} \ cdot D _ {\ text {iv}}} {AUC _ {\ text {iv}} \ cdot D _ {\ text {po}}}}} | 0,8 |
| Колебания | Пиковые колебания минимума в пределах одного интервала дозирования в установившемся режиме. | %пТF{\ displaystyle \% PTF} | %{\ displaystyle \%} |
CМаксимум,SS-Cмин,SSCсредний,SS⋅100%{\ displaystyle {\ frac {C _ {{\ text {max}}, {\ text {ss}}} — C _ {{\ text {min}}, {\ text {ss}}}} {C _ {{\ текст {av}}, {\ text {ss}}}}} \ cdot 100 \%}
|
41,8% |
Действие на вредные организмы
Дикват, как и другие производные дипиридилия, оказывают контактное неизбирательное действие на растения. Соединение относится к контактным гербицидам, разрушающим мембрану. В местах попадания на растения препарат разрушает ткани и быстро (в течение 2-4 суток) вызывает гибель растений. Характеризуется быстрым гербицидным эффектом и уничтожает надземную часть растений даже при использовании малых доз, используется также как дефолиант и десикант.
Гербицид не поглощается застаревшей корой, поэтому им можно обрабатывать приствольные круги ягодных, плодовых и цитрусовых культур. На свету при определенных условиях и относительной высокой влажности (100%) дикват может передвигаться по ксилеме, что ускоряет гибель надземной части растений.
Механизм действия
гербицидыгербицид
Передвижение производных дипиридилия по растению зависит от условий освещения и применяемых доз. При ярком освещении дипиридилиевые гербициды слабо передвигаются из обработанных листьев в корни в связи с прекращением оттока ассимилянтов, вызванным ингибированием процесса фотосинтеза. Уничтожение корней в этом случае не происходит. При слабой освещенности или в тени гербицидная активность сохраняются дольше, и токсические симптомы проявляются более равномерно.
Выведение лекарств из организма
ЛС и их метаболиты выводятся из организма разнообразными путями: с мочой, калом, желчью, секретом потовых, сальных и бронхиальных желез, молоком матери, воздухом, выдыхаемым воздухом.
Базовую роль в экскреции лекарств играют почки. На выведение лекарств влияют фильтрация, канальцевая реабсорбция и секреция. Фильтрации в клубочках нефрона испытывают вода, глюкоза, аминокислоты, белки с молекулярной массой до 60000 и некоторые другие соединения. Не фильтруются фракции препаратов, связанные с белками плазмы. Скорость фильтрации зависит от интенсивности кровообращения в почках.
Выделение лекарств с мочой
Активная секреция лекарственных средств происходит в проксимальных отделах нефрона. Секреция из крови через канальцевый эпителий в первичную мочу происходит с затратой энергии с участием специальных транспортных систем. Секретироваться могут как свободные, так и связанные с белками лекарственные средства. Реабсорбция лекарств происходит в дистальных отделах канальцев. Поскольку пассивная реабсорбция происходит через липидные мембраны канальцевого эпителия, то становится очевидным, что лучше реабсорбируются недиссоциированные липофильные молекулы слабых кислот и щелочей, а также нейтральные соединения. Степень реабсорбции зависит от рН мочи. Так, при кислых рН мочи слабые кислоты (барбитураты, бензодиазепины, сульфаниламиды) мало диссоциированные и легко реабсорбируются в кровь.
Напротив, в кислой среде молекулы слабых оснований (морфин, атропин, хинин и др.) находятся в высокодиссоциированном состоянии и плохо реабсорбируются в кровь, что способствует их выведению из организма. Регуляция рН мочи может быть использована при передозировках и отравлениях. Так, искусственно наполняя мочу с помощью гидрокарбоната натрия, можно повысить скорость вывода лекарств — слабых кислот. При отравлениях алкалоидами, которые по природе слабые основания, мочу необходимо подкислить. Вывод лекарств и различных метаболитов значительно замедляется у пациентов с почечной недостаточностью. Таким пациентам обычно назначают препараты, которые максимально метаболизируется в печени без образования активных метаболитов.
Выделение лекарств с калом
С калом выводятся из организма препараты, которые плохо всасываются в желудочно-кишечном тракте. Такие препараты используют преимущественно для воздействия на микрофлору кишечника или как слабительные средства.
Некоторые препараты (тетрациклин, пенициллины и др.) выделяются с желчью в тонкий кишечник, откуда они могут выводиться с калом или повторно всасываться, а затем снова выделяться в кишечник (так называемая циркуляция по энтеропеченочную кругу).
Другие способы выведения лекарств из организма
- Через легкие выводятся из организма летучие соединения. Этот процесс происходит за счет пассивной диффузии и зависит от частоты и глубины дыхания.
- Некоторые препараты выводятся с секретом желез (потовых, слюнных, желудочных и др.).
- Некоторые алкалоиды и основы могут выделяться в полость желудка, откуда затем всасываются повторно. При отравлении такими средствами проводят многократное промывание желудка, что позволяет удалить из организма определенное количество препарата.
- Вывод с секретом молочных желез (антикоагулянтов, транквилизаторов, цитостатиков и др.) создает опасность неблагоприятного воздействия лекарственных средств на организм ребенка.
Период полураспада — это просто
Употребление алкоголя в больших количествах сократит это время. Это было использовано для обеззараживания людей, которые подверглись внутреннему загрязнению тритиевой водой (тритием). Выведение этанола (алкоголя) из организма через окисление алкогольдегидрогеназой в печени ограничено. Например, концентрация алкоголя в крови может быть использована для изменения биохимии метанола и этиленгликоля. Таким образом окисление метанола до токсичныхформальдегида и муравьиной кислоты в организме может быть предотвращено приёмом соответствующего количества этанола человеком, употребившего метанол.
Снобы могут отметить, что как раз время распада таки зависит от наличия рядом таких же распадающихся атомов, ведь на этом принципе работает ядерная бомба и реактор. Стационарная концентрация лекарства в плазме крови — та концентрация, которая содержится в ней при поступлении препарата в организм с постоянной скоростью. В большинстве случаев R рассчитывается, используя показатели Css(max) и Сi(mах).
Биологи пытаются представить себе, как они функционируют в организме. Процессы взаимодействия низкомолекулярных лекарств с геном квалифицируются как фармакогеномика.

Работа генов определяет какие белки синтезируются в клетке, а от их разнообразия и активности зависят многие процессы, происходящие в организме. Отсюда еще одно направление в биологии, имеющее непосредственное отношение к фармакогенетике — протеомика, изучающая полный набор белков организма. Она связана с интенсивным изучением наследственных дефектов ферментных систем, выявляемых при применении лекарств.
Такое влияние может носить как общий, так и частный характер. Чувствительность по отношению к лекарствам меняется в зависимости от возраста. Для пациентов моложе 14 лет и старше 65 лет в силу возрастных особенностей организма отдельно устанавливают дозировки и частоту приема лекарств. Воздействие лекарства на организм, то есть его фармакодинамические свойства, практически не зависят от возраста пациента. Поэтому специальных лекарств для пожилых людей или для детей не существует.
Макролиды
Несмотря на то, что все макролиды в основе своей химической структуры имеют макроциклическое лактонное кольцо, их свойства, в том числе и фармакокинетические, значительно разнятся. Особенно выделяется в ряду макролидов азитромицин, содержащий дополнительно молекулу азота в макролидном кольце, что придает последнему повышенную устойчивость . Ключевые фармакокинетические свойства марколидов:
- Быстро всасываются из ЖКТ;
- Имеют невысокую биодоступность: кларитромицин и рокситромицин — 50 %, азитромицин — 37 %. Для последнего характерен эффект «первого прохождения» через печень — препарат частично инактивируется еще до поступления в системный кровоток;
- На всасывание некоторых макролидов влияет пища: она существенно снижает биодоступность эритромицина, в меньшей степени азитромицина и практически не влияет на биодоступность кларитромицина и спирамицина, хотя и замедляет их абсорбцию;
- Эритромицин нестабилен в солянокислой среде желудка, поэтому должен вводиться в составе солей, сложных эфиров или в форме таблеток с кишечнорастворимой оболочкой;
- Добавление метильной группы к эритромицину приводит к образованию кларитромицина, а присоединение к тому же эритромицину метилированного азота позволяет получить азитромицин. И кларитромицин, и азитромицин стабильны в солянокислой среде желудка и очень хорошо всасываются при пероральном применении;
- Период полувыведения эритромицина, кларитромицина и азитромицина составляет, соответственно, 1,5 часа, 6 часов и 68 часов. Таким образом, кратность применения эритромицина составляет 4 раза в день, кларитромицина — 2 раза в день, а азитромицина — 1 раз в день. При назначении кларитромицина в больших дозах возможно его применение 1 раз в день;
- Из-за большой продолжительности действия азитромицина 5‑дневный пероральный курс лечения с кратностью применения 1 раз в день считается адекватным при большинстве чувствительных к антибиотику инфекций;
- Все макролиды хорошо проникают в органы и ткани и считаются тканевыми антибиотиками. Пиковая концентрация в сыворотке крови намного ниже, чем в тканях (миндалины, придаточные пазухи носа, легкие, предстательная железа);
- Макролиды проникают внутрь клеток, создавая там высокие концентрации, что позволяет применять их для лечения внутриклеточных инфекций;
- Макролиды не проникают в спинномозговую жидкость. Поэтому их не применяют для лечения инфекций нервной системы;
- Проникают через плаценту и экскретируются в грудное молоко;
- Эритромицин и кларитромицин — важные ингибиторы фермента CYP450. При совместном применении с препаратами, метаболизирующимися в печени при участии ферментов цитохрома P450 (варфарин, другие непрямые антикоагулянты, карбамазепин, теофиллин, циклоспорин, алкалоиды спорыньи и другие), может усиливать эффект последних;
- Азитромицин не является ингибитором ферментов CYP450 и, соответственно, не вступает в лекарственные взаимодействия с препаратами, метаболизирующимися изоферментами цитохрома Р450;
- Выделяются с желчью;
- При почечной недостаточности период полувыведения не меняется, при печеночной — значительно увеличивается. Следовательно, в первом случае терапевтические дозы не корректируются, а во втором — снижаются.
Период полувыведения лекарства это: возраст и лекарства – Помощь Медика

Когда врачи выписывают рецепты на лекарства, они не просто записывают название лекарства на маленькой синей блокноте и отправляют своих пациентов в аптеку.
В рецепт включены подробности о том, сколько лекарства нужно принимать за один раз (доза) и через какие интервалы.
Эти инструкции, которые очень важны для обеспечения того, чтобы лекарство было эффективным и безопасным, частично основаны на периоде полувыведения назначаемого лекарства.
Как пациент, редко требуется знать период полураспада лекарственного препарата, который ваш врач хочет, чтобы вы принимали, но он может помочь понять, что означает этот термин и как он может повлиять на вас во время приема препарата.
Half-Life Defined
Биологический период полувыведения лекарства означает просто, сколько времени требуется для того, чтобы половина дозы метаболизировалась и выводилась из кровотока. Или, другими словами, период полураспада лекарства – это время, которое требуется для его сокращения вдвое.
Например, период полураспада ибупрофена (активного ингредиента в фирменных обезболивающих средствах, таких как Advil и Motrin) составляет около двух часов. Это означает, что если вы принимаете типичную дозу ибупрофена 400 мг (например, в полдень), половина дозы, 200 мг, будет выведена из вашего кровотока к 14:00. К 16:00 100 мг будет удалено, и так далее.
Важно отметить, что ожидаемый период полураспада лекарства будет варьироваться от человека к человеку, в зависимости от таких факторов, как возраст, вес, генетика и даже конкретные проблемы со здоровьем. Например, период полураспада ацетаминофена (активного ингредиента в Tylenol, другом ненаркотическом обезболивающем препарате) может значительно зависеть от функции печени человека, так как ацетаминофен в основном перерабатывается через печень
Например, период полураспада ацетаминофена (активного ингредиента в Tylenol, другом ненаркотическом обезболивающем препарате) может значительно зависеть от функции печени человека, так как ацетаминофен в основном перерабатывается через печень.
Интересно, что независимо от того, каков период полураспада лекарственного препарата, требуется приблизительно в четыре раза больше времени, чтобы концентрация лекарственного средства достигла устойчивого состояния в организме.
Это означает, что если вы начнете принимать лекарство с периодом полураспада 24 часа, через четыре дня или на пятый день, скорость приема препарата будет примерно равна скорости выведения.
Если период полураспада составляет 12 часов, вы достигнете устойчивого состояния в начале третьего дня (через 48 часов).
Почему период полураспада имеет значение
Препараты с более длительным периодом полувыведения требуют больше времени для работы, но, с другой стороны, им требуется меньше времени, чтобы покинуть кровоток.
С другой стороны, те, у кого короткий период полураспада, начинают действовать быстрее, но от них труднее отказаться.
На самом деле, лекарства с очень коротким периодом полураспада могут привести к зависимости, если принимать их в течение длительного периода времени.
Период полувыведения препарата является важным фактором, когда пора прекратить его прием. Будут рассмотрены как сила, так и продолжительность приема лекарства, а также период его полураспада
Это важно, потому что вы рискуете получить неприятные симптомы отмены, если бросите холодную индейку
Побочные эффекты от лекарств возникают обычно, когда уровень лекарственного средства в крови не находится в стабильном состоянии
Вот почему так важно следовать рекомендациям по дозировке и продолжительности приема
В противном случае организм будет реагировать, и действие препарата будет либо токсичным, как в случае, чем предполагалось, либо не терапевтическим, так как неэффективным для лечения.
Одно влияние периода полувыведения обнаруживается в антидепрессантах СИОЗС. Люди, принимающие СИОЗС с коротким периодом полураспада, гораздо чаще испытывают синдром отмены СИОЗС. Людям, принимающим СИОЗС с длительным периодом полураспада, таким как Прозак, нужно гораздо дольше ждать между прекращением приема Прозака и началом приема нового антидепрессанта, такого как ИМАО.
Период выведения и время полураспада лекарств
После всасывания в кровь лекарственные средства (ЛС) неравномерно распределяются в органах и тканях организма. Существенно влияют на распространение веществ биобарьеры. К ним относятся стенка капилляров, цитоплазматический, гематоэнцефалический (ГЭБ) и плацентарный барьеры.
Фторхинолоны
Все хинолоны имеют ряд общих фармакокинетических свойств :
Хорошо всасываются из ЖКТ и проникают в органы и ткани, создавая в них терапевтические концентрации. Пероральным формам отдается предпочтение перед парентеральными (при условии переносимости первых);
Биодоступность может достигать 99 %;
Наиболее высокие концентрации в тканях создаются при приеме офлоксацина и ломефлоксацина. Наименее высокие характерны для норфлоксацина;
Пища может замедлять процесс всасывания из пищеварительного тракта, но не влияет на биодоступность фторхинолонов;
Ципрофлоксацин, офлоксацин и пефлоксацин проникают через гематоэнцефалический барьер;
Все фторхинолоны проникают внутрь клетки и применяются для лечения внутриклеточных инфекций;
Большинство фторхинолонов выводятся почками, поэтому при нарушении их функций необходима корректировка дозы
Исключение составляет моксифлоксацин, который метаболизируется в печени и применяется при выраженной печеночной недостаточности с осторожностью.
Итоги
В заключении хотелось бы подчеркнуть — фармакокинетика часто кажется областью сложной и запутанной. Однако на деле фармакокинетические свойства в пределах одного класса препаратов зачастую схожи, и, выстроив для себя стройный ряд «биодоступность-всасываемость-элиминация» для конкретной группы, можно научиться быстро и легко отвечать на вопросы посетителя о том, когда и как принимать тот или иной антибиотик. Главное — желание и настойчивость, а их провизорам и фармацевтам не занимать.
Источники
- Bardal S. K., Waechter J. E., Martin D. S. Applied pharmacology. – Elsevier Health Sciences, 2011.
- Джекобс М. Новые подходы к оптимизации антимикробной терапии инфекций дыхательных путей с использованием фармакокинетических/фармакодинамических параметров //Клин микробиол антимикроб химиотер. – 2004. – Т. 6. – №. 1. – С. 22-32.
- Клиническая фармакология и фармакотерапия: учебник. – 3-е изд., доп. и перераб. / под ред. В.Г. Кукеса, А.К. Стародубцева. – М.: ГЭОТАР-Медиа, 2012. – 832 с.
- Morris Brown, Peter Bennett. Clinical Pharmacology 11th Edition. Elsevier Health Sciences, 2012.






